June 28, 2023

In Vitro Diagnostics (IVDs) are an essential part of healthcare, helping clinicians screen, diagnose, monitor, and treat various conditions. The In Vitro Diagnostic Regulation (IVDR) aims to improve the quality, safety, efficacy, and performance of IVD devices by increasing the requirements for their evaluation, regulation, and surveillance, thus significantly impacting clinical trials1-7.
The IVDR, introduced by the European Union (EU), came into effect in May 26, 2022 and replaced the previously established In Vitro Diagnostic Directive (IVDD)1,3. With increased regulatory oversight, the IVDR encompasses more stringent requirements for the designation of Notified Bodies (NBs) and monitoring by the national competent authorities and the Commission1–5. Thus, IVDR reduces the risks of discrepancies in interpretation across the EU market and enables timely detection of safety issues via post-market traceability and transparency. Unlike the IVDD, the IVDR places greater emphasis on product-life cycle management and continuous product evaluation. Manufacturers are obligated to demonstrate the implementation of an effective quality management system (QMS)5.
Successful IVD development requires a knowledgeable team. At CellCarta, we have the expertise to navigate the process of IVD development and provide quality and regulatory solutions for IVDR challenges.
How does IVDR impact clinical trial assays?
Within a clinical trial, all assays with a medical purpose are considered an IVD and are subject to IVDR3. More particularly, clinical trial assays used for medical management decisions of European subjects within the trial, involving patient selection, treatment allocation, and safety monitoring fall under IVD regulation (Figure 1). Thus, it is crucial to have a comprehensive understanding of the legislative requirements, and the devices affected, prior to initiating clinical trials. In-depth IVDR expertise minimizes the need for remediation following initial submission for performance evaluation and reduces the risk of commercial product withdrawal.
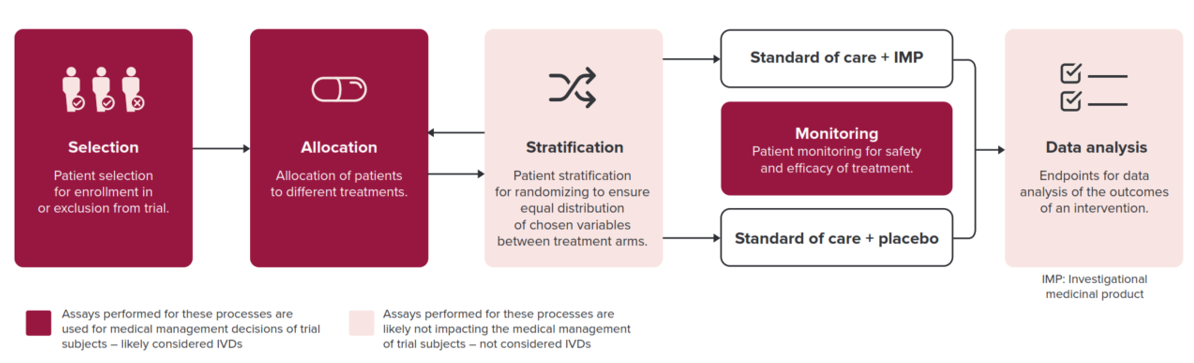
Figure 1. Processes determining which clinical assays are deemed IVDs and thus subject to IVDR. Exploratory assays for which correlation with clinical parameters is investigated retrospectively without having an impact on patients’ treatment are not subject to IVDR.
The shift from IVDD to IVDR brings about increased scrutiny of the performance evaluation plan concerning scientific validity, analytical performance, and clinical performance data5. The IVDR requires generation of clinical evidence to demonstrate conformity for all IVD devices, including those that were previously exempted under the IVDD3-5. As follows, clinical trial assays will need to provide more detailed and robust data on the performance of IVDs, including sensitivity, specificity, and predictive values2,4.
Additionally, IVDR introduces a new classification system for IVD devices, which considers the risks associated with the use of the device5. Some IVD devices previously classified as low-risk may now be classified as medium or high-risk and will require more extensive clinical trial data to obtain regulatory approval1,2,3,5.
The European Database on Medical Devices (EUDAMED) is a key aspect of rules established for IVDs under IVDR and is intended to improve transparency, traceability, and coordination among regulatory authorities and economic operations within the EU7. It enables efficient monitoring of devices throughout their lifecycle, enhances post-market surveillance, and strengthens patient safety.
What are Companion Diagnostics (CDx) devices?
Companion diagnostic (CDx) devices are a specific category of In Vitro Diagnostics that provide essential information for the safe and effective use of corresponding therapeutic products. CDx devices are categorized as Class C, representing a significant level of inherent risk6. At CellCarta, we specialize in providing comprehensive expertise in assay development and validation, guiding you through the transition of your assay from investigational clinical trial assay to a fully compliant IVD CDx.
Common questions, expert responses!
Is IVDR applicable for clinical trial testing in US-based laboratories?
- IVDR is applicable whenever testing occurs on EU patient samples, irrespective of laboratory location. IVDR is applicable when a testing laboratory employs clinical trial assays developed in-house or utilizes a CE-IVD kit for off-label use, provided that the test results serve a medical purpose.
Do clinical trials initiated before May 26, 2022 require a performance study application under IVDR?
- No, based on competent authority feedback, a performance study application is not required under IVDR if study subjects consented to participation in the trial before May 26, 2022.
Are all assays used in a clinical trial subject to IVD legislation?
- No, exploratory assays not intended for medical management decisions of patient care or treatment, are not subject to IVDR. These include stratification and endpoint analysis, or other exploratory assays, for which correlation with clinical parameters is investigated retrospectively without impact on patient treatment (medical purpose).
CellCarta’s team of quality, regulatory and assay development experts understand the challenges of IVD CDx development and work with you to implement effective strategies.
Contact us for guidance through the complex IVDR landscape and to receive personalized support for your clinical trial projects.
About the author:

Sarah Berwouts is a Director Quality at the CellCarta Antwerp site, leading the ISO 13485 implementation for the development of Histopathology and Genomics assays. Sarah has over 20 years of professional experience, including 8 years in medical laboratory quality systems, and coordination of the European proficiency testing scheme for cystic fibrosis. She has an 8 additional years of expertise in the development and manufacturing of IVDs at Multiplicom and Agilent Technologies, in different roles in R&D and Quality. Sarah has contributed to the implementation, maintenance, and harmonization of quality management systems under ISO 13485, ISO 15189, and ISO 17025. She studied Biomedical Sciences and received her PhD in quality improvement in medical laboratories.
References
- Lex – 32017R0746 – en – EUR-lex. EUR Available at: https://eur-lex.europa.eu/eli/reg/2017/746/oj. (Accessed: 26th June 2023)
- What’s changed compared to the IVDD – The European Union In Vitro Diagnostics Regulation. https://euivdr.com/whats-changed/.
- MDCG 2022-10 Q&A on the interface between Regulation (EU) 536/2014 on clinical trials for medicinal products for human use (CTR) and Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR). (2022).
- Dombrink, I. et al. Critical Implications of IVDR for Innovation in Diagnostics: Input From the BioMed Alliance Diagnostics Task Force. Hemasphere 6, E724 (2022).
- Factsheet for manufacturers of in vitro diagnostic. https://health.ec.europa.eu/system/files/2020-09/ivd_manufacturers_factsheet_en_0.pdf.
- Valla, V. et al. Companion Diagnostics: State of the Art and New Regulations. Biomark Insights 16, (2021).
- EUDAMED database – EUDAMED. https://ec.europa.eu/tools/eudamed/#/screen/home.